 |
| Dr. Howard Chang |
This is a case of a 12-year-old male with cerebral palsy, severe
developmental delay (level 1-2 years), and seizures (stable, no seizure
episodes since 2 years). He had progressive decline in neurological functions
following flu-like illness. He received IVIG and steroids for clinical
diagnosis of GBS-CIDP (18 months prior to death). Initially he showed some
improvement, but neurological functions continued to decline, with multiple
hospitalizations. MRI imaging studies (2 weeks prior to death) showed
extensive abnormal signal of the cerebral and spinal white matter. He was made
DNR. A general autopsy including brain and spinal cord was performed.
General Autopsy:
1. Atrophy of low
extremity muscles and apparent atrophy of muscles of hands.
2. Cushingoid
appearance with central obesity, skin striations, and adrenocortical atrophy
(likely due to steroid therapy).
Neuropathology Autopsy:
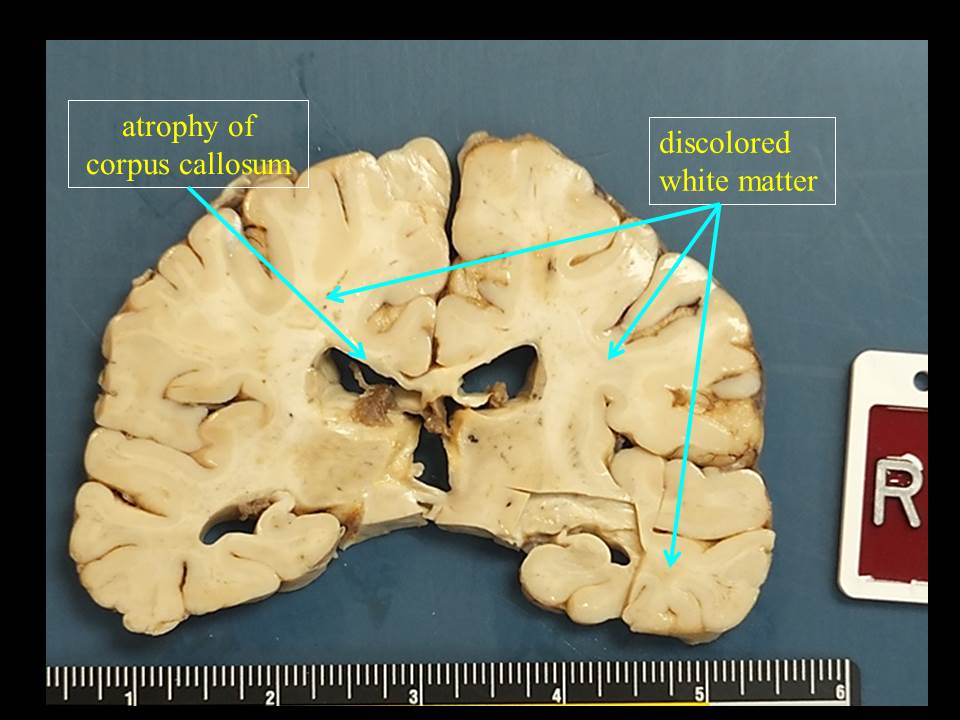
1. Extensive white matter atrophy-degeneration involving both the brain and spinal
cord (leukoencephalomyelopathy) with:
A. Microcephalic brain
(weight 1050 gm, normal should be about 1400 gm).
B. Bilateral cerebral
white matter atrophy-degeneration, with extensive astrogliosis and loss of
axons and myelin affecting the corpus callosum, and multifocal perivenous
microcystic changes involving the centrum semiovale, subcortical white matter,
with focal axonal spheroids in some of the microcystic areas.
C. Spinal cord with
extensive microcystic degeneration of white matter tracts with loss of axons
and myelin, affecting bilateral posterior, anterior and lateral columns.
Focal loss of neurons within the spinal cord gray matter is noted, including
the anterior horn motor neurons and those in the Clarke’s nuclei. The nerve
roots appear relatively unremarkable.
D. Increased
perivascular macrophages are noted within the brain and spinal cord sections,
but there are no other areas of significant inflammation involving the brain or
spinal cord parenchyma, or the nerve roots. There is no obvious evidence
of abnormal cytoplasmic inclusions within either the neurons or glia.
2. Cerebral infarcts, small, involving the right occipital pole (subacute), and a
lacunar (old) infarct superior to the right occipital horn of the lateral
ventricle.







1 comment:
Hi Karin,
Thank you very much for the email!
A full autopsy was performed, and the findings were relatively unremarkable, as noted in the blog post. The peripheral nerves were not specifically examined, but the both anterior and posterior nerve roots look rather unremarkable.
You are very perceptive to see that the hippocampi appear smaller than normal. The dentate gyrus do have fewer granule cells than normal, but I am not sure how is that related to "cerebral palsy" or the patient's leukoencephalomyelopathy.
A POLG-related mitochondria-depletion disorder is certainly a good possibility. Some of my differential diagnoses include Vanishing White Matter disease (due to mutations of the eukaryotic translation initiation factor 2B, eIF2B) or Leukoencephalopathy with Brainstem and Spinal Cord involvement and elevated Lactate (LBSL, due to mutation of the DARS2 gene that codes mitochondrial aspartyl-tRNA synthetase).
It turns out that the patient had a muscle biopsy before DNR, and some of the fresh muscle were saved frozen. We are waiting for parents' consent for genetic testing of some of the frozen tissue.
Karin Mente, MD:
I'm wondering if this could a mitochondrial DNA depletion disorder, such as a POLG related disorder. The patient that you've described doesn't seem to fit one particular POLG related disorder, but the pathological and clinical phenotypes are diverse because of different mutations leading to disease. Here's a more clinical review: https://www.ncbi.nlm.nih.gov/books/NBK26471/. I have yet to find a comprehensive review of POLG related neuropathology although I did look at some case reports.
I have a few questions about your patient. What was the extent of the non-CNS autopsy? Were there any hepatic or cardiac gross and microscopic changes? In peripheral nerve, was there axonal loss and/or demyelination? Looking your gross brain slice image -- the hippocampi appear atrophic. Is that correct? I'm also curious about the "cerebral palsy."
As far as how to proceed with this case -- whole exome sequencing. Although it is possible to recover genomic DNA from FFPE, I've been told that it is difficult so using frozen tissue or blood would be much more efficient. WES is quite expensive and any possibly or potentially damaging variants would need to be confirmed with Sanger. and then if the variants haven't been reported as mutations associated with disease, then they are compared with WES from unaffected first degree relatives, usually parents.
Post a Comment